الثلاثاء، 8 مارس 2011
Effects of Thalassemia
It can result in bigger, thin and brittle bones.
It can also make people prone to developing all sorts of infections
It can lead to heart rhythm disorders, also known as arrythmias. (dizziness, sweating, skipping heartbeats and pounding heartbeats )
It can also lead to an enlarged spleen
Alpha (α) thalassemias
The α thalassemias involve the genes HBA1and HBA2, inherited in a Mendelian recessive fashion. There are two gene loci and so four alleles. It is also connected to the deletion of the 16p chromosome. α thalassemias result in decreased alpha-globin production, therefore fewer alpha-globin chains are produced, resulting in an excess of β chains in adults and excess γ chains in newborns. The excess β chains form unstable tetramers (called Hemoglobin H or HbH of 4 beta chains) which have abnormal oxygen dissociation curves.
Beta (β) thalassemias
Beta thalassemias are due to mutations in the HBB gene on chromosome 11 , also inherited in an autosomal-recessive fashion. The severity of the disease depends on the nature of the mutation. Mutations are characterized as (βo or β thalassemia major) if they prevent any formation of β chains (which is the most severe form of β thalassemia); they are characterized as (β+ or β thalassemia intermedia) if they allow some β chain formation to occur. In either case there is a relative excess of α chains, but these do not form tetramers: rather, they bind to the red blood cell membranes, producing membrane damage, and at high concentrations they form toxic aggregates.
Delta (δ) thalassemia
As well as alpha and beta chains being present in hemoglobin about 3% of adult hemoglobin is made of alpha and delta chains. Just as with beta thalassemia, mutations can occur which affect the ability of this gene to produce delta chains.
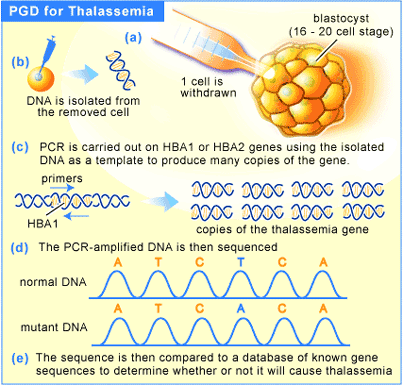
Thalassemia (also spelled thalassaemia) is an inherited autosomal recessive blood disease. In thalassemia the genetic defect, which could be either mutation or deletion, results in reduced rate of synthesis or no synthesis of one of the globin chains that make up hemoglobin. This can cause the formation of abnormal hemoglobin molecules, thus causing anemia, the characteristic presenting symptom of the thalassemias.
Thalassemia is a quantitative problem of too few globins synthesized, whereas sickle-cell anemia (a hemoglobinopathy) is a qualitative problem of synthesis of an incorrectly functioning globin. Thalassemias usually result in underproduction of normal globin proteins, often through mutations in regulatory genes. Hemoglobinopathies imply structural abnormalities in the globin proteins themselves. The two conditions may overlap, however, since some conditions which cause abnormalities in globin proteins (hemoglobinopathy) also affect their production (thalassemia). Thus, some thalassemias are hemoglobinopathies, but most are not. Either or both of these conditions may cause anemia.
The two major forms of the disease, alpha- and beta- (see below), are prevalent in discrete geographical clusters around the world - probably associated with malarial endemicity in ancient times. Alpha is prevalent in peoples of Western African descent, and is nowadays found in populations living in Africa and in the Americas. Beta is particularly prevalent among Mediterranean peoples, and this geographical association was responsible for its naming: Thalassa (θάλασσα) is Greek for the sea, Haema (αἷμα) is Greek for blood. In Europe, the highest concentrations of the disease are found in Greece, coastal regions in Turkey, in particular, Aegean Region such as Izmir, Balikesir, Aydin, Mugla and Mediterranean Region such as Antalya, Adana, Mersin, in parts of Italy, in particular, Southern Italy and the lower Po valley. The major Mediterranean islands (except the Balearics) such as Sicily, Sardinia, Malta, Corsica, Cyprus and Crete are heavily affected in particular. Other Mediterranean people, as well as those in the vicinity of the Mediterranean, also have high rates of thalassemia, including people from the West Asia and North Africa. Far from the Mediterranean, South Asians are also affected, with the world's highest concentration of carriers (16% of the population) being in the Maldives.
The thalassemia trait may confer a degree of protection against malaria, which is or was prevalent in the regions where the trait is common, thus conferring a selective survival advantage on carriers (known as heterozygous advantage), and perpetuating the mutation. In that respect the various thalassemias resemble another genetic disorder affecting hemoglobin, sickle-cell disease.
الاشتراك في:
الرسائل (Atom)